Objectives of the Presentation
 Describe the clinical characteristics of canine hereditary ataxias using American Staffordshire terriers, Old English sheepdogs and Scottish terriers as examples.
Describe the clinical characteristics of canine hereditary ataxias using American Staffordshire terriers, Old English sheepdogs and Scottish terriers as examples.
Discuss current understanding of the genetic basis of human hereditary ataxia.
Explain the approach to identification of the genetic cause of hereditary neurodegenerative diseases including evaluation of candidate genes, linkage analysis and, briefly, association studies.
Describe the linkage studies performed in two breeds of dog highlighting the potential problems with each step.
Overview of the Issue
The Cerebellum
The cerebellum is a complex part of the brain that controls the range, rate and force of movements, and as such is vital to an animal's ability to move. Cerebellar diseases manifest as incoordination, intention tremors and lack of balance and in spite of preservation of strength, severe cerebellar disease is incapacitating. The cerebellum is comprised of a three-layered cortex and a central core of nuclei. The external cortical layer of neurons is called the molecular layer, a thin layer of Purkinje cells that provide the majority of the output from the cerebellar cortex lie below the molecular layer and deep to the Purkinje cells there is a thick layer of small neurons called granule cells. The axons to and from these neuronal layers course through centrally located white matter to a series of nuclei located at the core of the cerebellum. The cerebellum, in particular the Purkinje cells, is sensitive to insult and is a common target of neurodegenerative diseases. There is a large group of neurodegenerative disorders that affect the cerebellar cortex primarily called the cerebellar abiotrophies1. Cerebellar abiotrophy is a collective term that implies a hereditary intrinsic defect that causes progressive neuronal death as the animal ages. The term abiotrophy is not commonly used in human medicine, and this group of diseases can also be referred to as hereditary ataxia, spinocerebellar ataxia or cerebellar cortical degenerative diseases.
Canine Hereditary Ataxia
Cerebellar degenerative diseases have been described in a large number of breeds of dog, but most of these reports are descriptions of single animals. However, there are a number of cerebellar disorders that have been shown to be inherited and are posing a significant problem to the breeds involved. These breeds include the Gordon Setter2, the Brittany spaniel3, the Old English sheepdog4, the American Staffordshire terrier5 and the Scottish terrier. Clinically this group of diseases causes insidious onset of ataxia characterized by dysmetria (abnormal range of movement), intention tremors (head tremors when the animal is focused on something), truncal sway (pronounced sway of the body from side to side when walking), spontaneous nystagmus (abnormal uncontrolled eye movements) and loss of physiologic nystagmus. Age of onset and rate of progression of signs varies between breeds. Signs are first noted at 6 to 40 months of age in the Gordon setter, and the Old English sheepdog, and as early as 8 weeks in the Scottish terrier. Signs appear much later in the American Staffordshire terrier and the Brittany spaniel, typically between 3 and 8 years of age. Signs progress slowly but at different rates depending on the breed affected, and ultimately the animal is unable to walk without falling, and has difficulty eating and drinking due to their intention tremor. Of the breeds listed above, it is unusual for Scottish terriers to be euthanized because of their cerebellar disease--while they have an unusual gait, it is not incapacitating. By contrast, the American Staffordshire terriers, even though they have later onset of signs, are usually euthanized because of their disease between 2 and 8 years from onset of signs. Old English Sheepdogs and Gordon Setters may experience stabilization of their signs, but some do progress until they need to be euthanized.
Diagnosis of these diseases antemortem is based on the presence of signs of cerebellar dysfunction on neurological examination in an otherwise healthy dog, a consistent history of chronic, progressive signs (possibly with related dogs affected), normal blood work and atrophy of the cerebellum on magnetic resonance images. Definitive diagnosis can be made at necropsy; atrophy of the cerebellum is visible grossly and histopathologically there is Purkinje cell degeneration followed by neuronal loss in the molecular and granular layers of the cerebellum. The histopathological changes are largely limited to the cerebellar cortex in the breeds we are investigating. To date, disease has been confirmed on necropsy in every dog that we have suspected to have hereditary ataxia based on clinical signs and history, and so a consistent history and clinical signs appears to be a reliable means of diagnosing the disease.
Pedigree analysis in the Gordon Setter,the Old English sheepdog,the American Staffordshire terrier and the Scottish terrier has shown the disease to be inherited by an autosomal recessive trait. This means that dogs have to have two copies of the mutant allele to show signs of disease. Dogs with only one copy of the mutant allele (a carrier) are clinically normal and breeders are unaware that they could produce affected dogs if bred to another carrier. The recessive nature of the diseases, and the late onset of signs in affected dogs in some breeds results in the unwitting widespread breeding of affected and carrier dogs, disseminating the deleterious allele within the breed. This is particularly true in the American Staffordshire terrier. In this breed, signs often do not appear until five years of age or older, and we estimated the deleterious allele to have a prevalence of 39% in the pedigrees we examined. Indeed, the problem has been recognized in dogs from all geographic areas of America and in Europe, and we continue to recruit affected dogs to our study at the rate of 1 dog a month, nearly 5 years after initially starting to work on the problem. Clearly, there is a real need for a genetic test to identify carrier and affected status in all 4breeds of dog.
Human Hereditary Ataxia6
Hereditary ataxias are a well-recognized group of devastating neurodegenerative diseases in humans with an estimated prevalence as high as 3 in 100,000 in certain populations. In humans, the term hereditary ataxia encompasses diseases in which there is cerebellar degeneration either alone, or in combination with spinal cord degeneration and/or a sensory neuropathy. The progressive neuronal degeneration results in signs very similar to those exhibited by affected dogs, namely insidious development of ataxia, difficulty speaking and abnormal eye movements, in addition to a number of other disease dependent neurological signs. Onset of deficits may occur in infancy, childhood or well into adulthood and signs progress at varying rates until the patient is incapacitated. As in dogs, the most common histopathological finding is progressive loss of neuronal populations within the cerebellum, specifically of the Purkinje neurons and the granular cell layer causing cerebellar atrophy that can be detected on magnetic resonance images. Neuronal loss from other areas of the central and peripheral nervous system may also occur dependent on the particular disease.
The complexity of this group of diseases in humans can present a very confusing clinical picture, and indeed, it is difficult to differentiate between them based on clinical findings alone. To resolve this problem, they are now categorized according to the mode of inheritance, and more recently according to the chromosomal locus or gene with which they are associated. Inheritance of ataxia can occur in an autosomal dominant or recessive fashion. More rarely, X-linked and mitochondrial inheritance have been reported. Autosomal dominant cerebellar ataxias (ADCA) are more common than their recessive counterparts, and are currently classified into 26 different types according to the gene or chromosomal locus associated with the disease. Ten of these disorders share an underlying genetic mutation mechanism of unstable expansions of trinucleotide (CAG or CTG) or pentanucleotide (AATTCT) repeats in untranslated or coding regions of the responsible genes. The number of different loci associated with ADCA expands each year, but in spite of this, the genetic cause of up to 60% of cases is unknown.The autosomal recessive cerebellar ataxias (ARCA) include a very diverse group of disorders7. The most common ARCA is Friedreich's ataxia, which is also caused by a trinucleotide expansion (GAA). Prevalence of this disease is approximately 1 in 50,000 people. Other forms in which a mutation has been identified include spinocerebellar degeneration caused by primary vitamin E deficiency, abetalipoproteinemia, ataxia with oculomotor apraxia types 1 and 2, ataxia telangiectasia, autosomal spastic ataxia of Charlevoix-Saguenay, Marinesco Sjogren, spinocerebellar ataxia with axonal neuropathy 1, and infantile onset spinocerebellar ataxia. There are many additional reports of families with different forms of ARCA for which a genetic locus has not been identified. Presenting clinical signs are extremely diverse in this group, with a wide variety of additional problems present reflecting the involvement of multiple systems. For example, Friedreich's ataxia causes a cardiomyopathy, and ataxia telangiectasia is associated with immune deficiency.
The discovery of causative mutations has allowed researchers to focus on potential mechanisms of neuronal death. The CAG trinucleotide repeats detected in six of the spinocerebellar ataxias have received particular attention. These CAG repeats result in the expression of polyglutamine (or polyQ) sequences, but the precise mechanism by which these sequences cause neuronal death is still unknown. A number of theories are proposed including direct cytotoxicity, interaction with transcription factors, alteration of calcium channel function, obstruction of axonal transport, impairment of proteolysis, misfolding of proteinsand phosphorylation abnormalities. Other processes known to be important factors in causing cell death in this group of diseases include mitochondrial function, DNA repair, and activity of fibroblast growth factor.
Finding the Gene
There are several different approaches that can be used to identify the abnormal gene responsible for inherited diseases. The first is to identify candidate genes by comparing the disease in dogs to other similar diseases for which the mutation is known in other species (e.g., human and rodent diseases). A good example of using this approach successfully was the identification of a mutation in the gene for dystrophin in golden retrievers by a direct comparison of their disease to humans with muscular dystrophy. Because of the similarities between the human and canine hereditary ataxias, the known human mutations can serve as candidate genes for the canine forms of the disease. There are also a large number of rodent models of cerebellar degenerative diseases that have been defined genetically and can provide candidate genes. However, the possible genome-wide list of candidate genes includes more than 50 genes, and as sequencing one gene in affected and normal dogs to identify a mutation can represent a year of work, a straightforward candidate gene approach is not practical in the hereditary ataxias.
The second approach is to perform linkage analysis. In this type of study, markers spaced at regular intervals throughout the canine genome are used to screen the genome. PCR reactions are performed using each of over 300 markers and each individual dog will produce slightly different length PCR products. These represent alleles and they are inherited through generations. By comparing the alleles of affected dogs and their relatives, it is possible to track the inheritance of a particular allele and to compare it to the inheritance of the disease, thus linking disease to the pedigree. The statistical comparison of the inheritance of each allele and the inheritance of the disease is called linkage analysis and produces a LOD score. LOD scores greater than 3 show that the chromosomal region in which that allele is located is linked to the disease. In order for linkage analysis to be successful, it is necessary to have DNA from multigenerational families of affected dogs and to know for sure whether or not they have the disease or are normal. Linkage analysis and candidate gene approaches can be combined by first performing linkage analysis in dog families to define a small chromosomal region linked to the disease, and then to identify candidate genes in this region for further investigation.
The final approach is to perform association studies. These studies use a 'SNP chip' to screen the canine genome at very high density for genetic differences that are associated with the disease. In order to do these studies it is necessary to have DNA from a large number of affected individuals and an equally large number of unrelated normal individuals. This can pose a problem as we do not always have DNA from large numbers of affected dogs when dealing with neurodegenerative diseases, and it can be difficult to get truly unrelated dogs from the same breed, due to the genetic nature of a canine breed. If successful it is a rapid way to screen animals for a causative mutation.
Linkage Studies on Canine Hereditary Ataxia
At NCSU we are performing linkage studies in American Staffordshire terriers, Old English sheepdogs and Scottish terriers. The first step in this process is to collect DNA (extracted from a blood sample) from families of affected dogs, to confirm whether dogs are affected or normal, and to collect pedigrees. It can take literally years to collect DNA from multiple generations of affected dogs families. The second step is to perform a power analysis. To do this a simulated linkage study is performed using the pedigree information collected on the dogs for which there is DNA. This step is very important because it allows us to determine whether we have collected enough DNA samples from families of affected dogs to be able to establish linkage (i.e., have we got adequate power within our families of dogs). Figure 1 illustrates the results of a simulation study. Genome screens are expensive and time consuming so it is important to demonstrate that there is a statistically good chance on a positive outcome. To do this type of 'power analysis' the statistician simulates the gene being linked to one of the markers by different distances and runs the software program that is used for the linkage studies. In all 3 breeds we were able to demonstrate that there was a good chance of establishing linkage.
| Figure 1. | 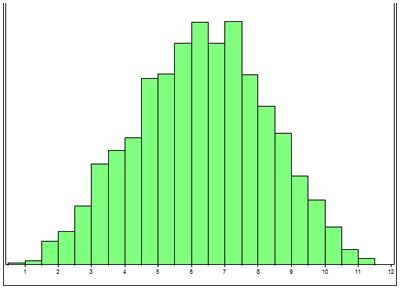
LOD score (x axis) vs number of simulations (y axis). The majority of the simulations generate a LOD score greater than 3. |
|
| |
The third step is to run the PCR reactions on all of the markers to be used in the initial genome screen and to analyze the results to determine the alleles for each marker in each individual. These allele 'calls' are then used by the statistician to perform the linkage analysis itself. Typically we start by screening 48 dogs and to date we have completed an initial genome screen and established linkage in the American Staffordshire terriers and Old English sheepdogs and we are awaiting funding to do the screen in Scottish terriers.
Once linkage has been established, a list of the genes known or suspected to be located in the linked area can be obtained from the canine genome assembly (available through the internet). If you are lucky, an excellent candidate gene is found in the linked region and can be sequenced for the presence of mutations. However, it is more common that the region contains upward of 50 genes and none are clear candidates. In this instance, the linked region is screened again by identifying markers at intervals along the region and repeating the linkage study with the hope of narrowing the region of interest. In order to this type of high density screening, the power of the families of dogs needs to be higher and more individuals need to be genotyped. This is ongoing in the American Staffordshire terriers and Old English sheepdogs. The aim of the linkage study is to identify a chromosomal region as being linked to the disease. It does not directly identify the mutant gene. This is done by sequencing genes that are linked to the disease.
Potential Pitfalls
There are numerous ways in which these studies can fail. The first is if the dogs are incorrectly identified either as being affected or normal or within a family. We do encounter occasional problems with dogs clearly not being parented by the dogs that are listed, but fortunately this is unusual. The second big hurdle is collecting blood samples from multiple generations of dogs. It is critical that samples can be obtained from 3 generations of dogs in order to generate adequate statistical power, but this can be difficult to do, particularly in adult onset diseases. Breeders usually play a crucial role in the success of sample collection (Figure 2).
| Figure 2. Pedigrees from two families of affected dogs. | 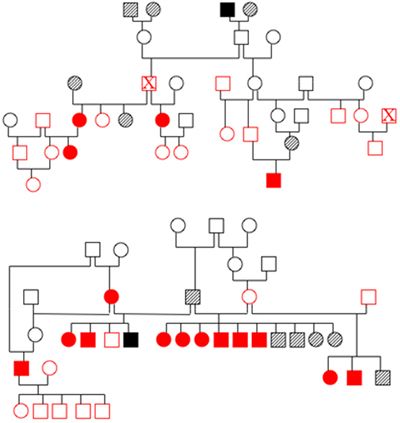
|
|
| |
Open symbols indicate dogs with a normal phenotype, solid symbols are affected dogs and hatched symbols are dogs of unknown phenotype. Red indicates that DNA has been banked. Both families are related to each other three generations above the dogs shown in the diagram. The individual marked by an X appears twice in pedigree 1. Identifying and obtaining samples from all of these dogs once they have dispersed to different owners represents a real challenge.
Once we have the DNA samples in hand, we can encounter problems with our genome screens. A particular marker will only give us information about a chromosomal region if the dogs being investigated have different alleles of that marker--if they all have the same allele, then we cannot distinguish between them and the marker is deemed uninformative. Pure bred dogs, by definition, are in bred and as a result, it is not uncommon to find markers are identical in all dogs of a family. This leaves holes in the chromosomal screen and can result in a false negative for linkage. Finally, once linkage has been established, if there is no good candidate gene, high density screening of the linked area requires even stronger families of dogs.
Summary
Hereditary ataxia is emerging as an important group of neurodegenerative diseases in dogs. The recessive nature of the disease and the late onset of signs in some breeds has resulted in wide dispersal of the causative mutation.
Linkage studies have been performed in American Staffordshire terriers and Old English sheepdogs to establish linkage and further refinement of the linked regions is ongoing. Work on the Scottish terriers will start soon.
The best way to control these diseases is to identify the underlying mutation and to develop a genetic test for the disease trait. Ongoing research into the mutation in several breeds of dog is showing promising results and we hope that genetic tests will become available to breeders in the near future.
References
1. de Lahunta A. Abiotrophy in domestic animals: a review. Am. J. Vet. Res. 1990;54:65-67.
2. de Lahunta A, Fenner WR, Indrieri RJ, et al. Hereditary cerebellar cortical abiotrophy in the Gordon Setter. J Am Vet Med Assoc 1980; 177:538-541.
3. Higgins RJ, LeCouteur RA, Kornegay JN, Coates JR. Late-onset progressive spinocerebellar degeneration in Brittany Spaniel dogs. Acta Neuropathol. 1998;96:97-101.
4. Steinberg HS, Van Winkle T, Bell JS, de Lahunta A. Cerebellar degeneration in Old English Sheepdogs. Am. Vet. Med. Assoc. 2000;217:1162-1165.
5. Olby NJ, Blot S, Thibaud J-L, Phillips J, O'Brien DP, Burr J, Berg J, Brown T, Breen M Cerebellar Cortical Degeneration in Adult American Staffordshire Terriers. J Vet Intern Med 2004;18:201-208.
6. http://www.genetests.org/ Go to gene reviews and then hereditary ataxia.
7. Di Donato S, Gellera C, Mariotti C. The complex clinical and genetic classification of inherited ataxias. II Autosomal recessive ataxias. Neurol. Sci. 2001;2:219-228.