Simon R. Platt, BVM&S, MRCVS, DACVIM (Neurology), DECVN
RCVS Specialist in Veterinary Neurology Head of Neurology/Neurosurgery Unit, Centre for Small Animal Studies, The Animal Health Trust, Kentford, Newmarket, Suffolk, England
Cerebrovascular accidents (CVA) are one of the major causes of disability among human adults. Previously considered uncommon, CVA are increasingly recognized in dogs or cats with the advances of neuro-imaging. Most types of CVA that are seen in humans have been documented in dogs.1 Recovery from cerebrovascular disorders in animals is probably more spectacular than in humans because animals have a less prominent pyramidal system.2 A 'stroke' is a suddenly developing focal neurological deficit resulting from a cerebrovascular accident.3 The causes of strokes can be divided into two basic groups: (1) obstruction of the blood vessels leading to ischemia, and (2) rupture of blood vessel walls leading to hemorrhage.4
The central nervous system (CNS) requires a continuous supply of glucose and oxygen to sustain its high expenditure of energy. The transportation of these fuel molecules requires sufficient blood flow through a cerebral vasculature with adequate capacity. In the dog, blood supply to the brain arises from the basilar and internal carotid arteries, which join at the base to form the arterial circle of Willis.5 The cerebrum is supplied by three pairs of cerebral arteries arising from this arterial circle, with each one responsible for the perfusion of large but overlapping areas of the cerebrum.5 Any diseases which affect the cerebral blood vessels will cause disturbances of the cerebral blood flow (CBF) which in turn can lead to tissue damage. The metabolism of the brain is solely aerobic and without any significant energy reserves. The exceptionally high demand for circulating blood and oxygen is reflected is reflected in the disproportionately high rate of CBF compared with flow to other parts of the body, comprising 20% of the cardiac output and 15% of oxygen consumption when the body is at rest, even though the brain makes up only 2% of the body weight.4
Cerebral ischemia is the reduction, although not necessarily the cessation, of blood flow to a level incompatible with normal function; the impairment may be global or regional.4,6 Ischemia, viewed simplistically as hypoxia plus hypoglycemia, will affect the most sensitive elements in the tissue, and if severe, persistent, or both, perturb all components. In its mildest form, impaired regional CBF causes a transient ischemic attack (TIA). TIA has an abrupt onset but is a rapidly diminishing neurological deficit of vascular origin, which lasts for less than 24 hours.4,5 This is well documented in humans but has not been studied in dogs, although the authors do believe that this occurs in dogs, occasionally as a historical precursor to an infarction.
Severe ischemia, which in the CNS would produce necrosis of the neurons and glial elements, results in an area of dead tissue termed an infarct.6 Severe arterial hypotension produces bilateral infarction in the boundary or watershed zones between major arterial territories.4 The critical threshold values of CBF needed for the maintenance of functional and structural integrity of the brain has been determined to be approximately 40% of the normal value (i.e., approx. 20 ml/100 g per minute).4 From about 40% to 30%, increasing numbers of neurons are unable to produce sufficient energy to maintain the functions needed for the transmission of nerve impulses, and at about 30% of normal blood flow transmission ceases completely although the cells can stay alive, as in a TIA. If regional CBF further diminishes below about 15% of normal (10-12 ml/100 g per minute), there is absolute membrane failure resulting in an irreversible nerve cell injury, as in an infarct.4 These levels can be higher in an already compromised brain.
In humans, there are regions of vulnerability within the brain where neurons are prone to be injured by global hypoxia-ischemia and hypoxia. These areas are the cerebral cortex, the hippocampus, the amygdala, several basal and thalamic nuclei, and the cerebellar cortical Purkinje cells.6
Infarction can result from arterial obstruction or venous thrombosis; arterial infarction can be due to either obstruction from thrombosis or embolism or to occlusion from blood vessel abnormalities such as vasculitis.5 A number of classification systems for ischemic stroke have been proposed in humans. The most commonly used clinical systems divide ischemic stroke into three major stroke subtypes: large artery or atherosclerotic infarctions, cardioembolic infarctions and small vessel or lacunar infarctions.7 Atherosclerotic infarctions are the most common subtype documented in people.7 Although the frequency of the three different subtypes is as yet unknown in dogs, atherosclerosis has been reported in dogs; it is especially seen in older dogs, dogs with hypothyroidism, and Miniature Schnauzers with idiopathic hyperlipoproteinemia.5 Other diseases associated with infarction in dogs include sepsis, coagulopathy, neoplasia and heartworm infections.5 The use of MR with techniques such as diffusion weighted imaging and angiography may well help to define the subtype of infarction in the future. Because of abundant venous anastomoses, venous infarction is uncommon in dogs; as arterial blood flow is preserved, hemorrhage and edema tend to be more severe in venous infarction than in arterial infarction.8
Cerebrovascular accidents can on occasion result from hemorrhage.5 This can occur within or around the brain and may result in rapid cerebral dysfunction often by alteration in cerebral volume (mass effect). It is classified as epidural, subdural, subarachnoid, intraparenchymal (primary or secondary), or intraventricular.5 When the bleeding is substantial enough to form an excessive additional volume within the CNS, the results can be fatal. The presence of a hematoma initiates edema and neuronal damage in surrounding parenchyma.9 Fluid begins to collect immediately in the region around the hematoma, and edema usually persists for up-to 5 days,9 and in some cases as much as 2 weeks.10 Early edema around the hematoma results from the release and accumulation of osmotically active serum proteins from the clot.9 Vasogenic edema and cytotoxic edema subsequently follow owing to the disruption of the blood-brain barrier, the failure of the sodium pump, and the death of neurons.11 The delay in the breakdown of the blood-brain barrier and the development of cerebral edema after intracerebral hemorrhage suggest that there may be secondary mediators of both neural injury and edema. It had been thought that cerebral ischemia occurred as a result of mechanical compression in the region surrounding the hematoma, but recent studies in animals and humans have not confirmed this.12 It is currently thought that blood and plasma products mediate most secondary processes that are initiated after an intracerebral haemorrhage.12 Neuronal death in the region around the haematoma is predominantly necrotic, with recent evidence also suggesting the presence of programmed cell death (apoptosis).12
The source of primary intraparenchymal hemorrhage is incompletely understood but human patients often have systemic hypertension with concurrent fibrinoid degeneration of arteries in the brain.13 Hypertension in dogs may be primary or secondary to disorders such as renal disease, and hyperadrenocorticism; these animals may be predisposed to intracranial hemorrhage.14 A variety of secondary causes of hemorrhage exist in dogs. Dogs with brain infarction can have associated hemorrhage, as can dogs with intracranial tumors, vasculitis or coagulopathies.5
Clinical Signs
CVA are characterised clinically by a peracute or acute onset of focal, asymmetrical and non-progressive brain dysfunction.5 Worsening of edema (associated with secondary injury phenomenon) can result in progression of neurological signs for a short period of 24-72 hours. Hemorrhage may be an exception to this description and be presented with a more progressive onset. Clinical signs usually regress after 24-72 hours; this is attributable to diminution of the mass effect secondary to hemorrhage and reorganisation or edema resorption.15 With brainstem involvement, neurological examination of the cranial nerves will define the exact location and extension of the lesion. With forebrain lesion, the clinical sign may vary from simple disorientation to death. A unilateral lesion will induce ipsilateral circling, hemi-inattention syndrome, contralateral central blindness, as well as contralateral ataxia and proprioception deficits. Seizures are reported to be very common in association with CVA in dogs.16
Diagnosis
Blood and urine analysis is indicated to identify the possible underlying causes described above. Thyroid function (FT4, TT4 and endogenous cTSH levels), a coagulation profile (including a buccal mucosal bleeding time, a prothrombin time, a partial thromboplastin time and fibrinogen degradation products), and if possible multiple systolic blood pressures and an ECG, should be evaluated in any animal suspected of CVA. A fecal analysis should be performed to rule out parasitic infestation. Blood and urine cultures are indicated in case of sepsis. Cerebrospinal fluid analysis is unlikely to confirm a diagnosis of CVA but may help to rule-out inflammatory CNS disease or may on occasion reveal recent haemorrhage (xanthochromia), normal to increased protein and a mild neutrophilic or mononuclear pleocytosis.5 Imaging studies of the brain (computed tomography {CT} or magnetic resonance imaging {MRI}) are necessary to confirm the clinical neurolocalisation, re-enforce the suspicion of CVA, identify associated mass effect and rule-out other causes of focal brain disorders (trauma, tumor, inflammation). CT also allows rapid image acquisition, in addition to the fact that changes associated with ischemia/infarction can be detected as early as 3 to 6 hours after the onset.7 Enhancement usually appears after 24-48 hours and is most evident after 1 or 2 weeks especially in the periphery where neovascularistion exists.17
MR imaging is more sensitive than is CT in early infarction, with changes seen within an hour if onset.18 Magnetic resonance imaging is more sensitive in the detection of edema, provides multiplanar views, and lacks beam-hardening artifact when compared with CT.7 The conventional imaging findings in evolving cerebral infarction are well characterized and follow a temporal evolution similar in many ways to that seen on CT.18 These changes seen in ischemic parenchyma rely on an increase in tissue water content.7 Gradually, during the acute stage, the T2-weighted image becomes more hyperintense in the ischemic region, particularly over the first 24 hours 7 These signal changes seen in the first 24-hours are best appreciated in grey matter and are well visualised in deep grey matter structures such as the thalamus or basal ganglia, in addition to cortical grey matter. Gadolinium enhances infarcts because of vascular rupture but does not enhance ischemia or edema.
Computed tomography is very sensitive for acute hemorrhage, with a linear relationship demonstrated between CT attenuation and hematocrit.19 In a patient with a normal hematocrit, acute hemorrhage is seen as an area of increased attenuation, which tends to increase for the first 72 hours and then slowly decreases to isodensity at about 1 month post-hemorrhage.19 The periphery of the lesion may enhance from approximately 6 days to 6 weeks after onset, on a CT scan.
The initial MRI appearance of haemorrhage is dependent on the age of the hematoma, among other determinants, which determines its unique signal intensity patterns.20 Localization of hemorrhage to the parenchyma or 'extra-axial' space is central to assessing the etiology and the initiation of treatment.20 In dogs, it is more common to see intraparenchymal than 'extra-axial' hemorrhage, the latter of which is typically subdural in location.
Other imaging modalities which may be utilised to investigate CVAs include; cerebral angiography, to demonstrate vascular malformations; cerebral scintigraphy as a non-specific way to identify a brain lesion; Doppler ultrasonography to analyse cerebral blood flow; and single photon emission computed tomography (SPECT) to analyse regional blood flow. These modalities are not frequently used now as the advances possible with MR technology mean that blood vessel abnormalities and regional blood flow can be assessed in conjunction with the structural abnormalities suggestive of a CVA.
Treatment and Prognosis
There is no specific treatment for infarctions and the majority of intraparenchymal hemorrhages. The treatment of any type of CVA focuses on maintaining cerebral perfusion, through maintenance of systemic blood pressure, and subsequent tissue oxygenation, as well as the management of secondary neurologic sequelae such as seizures, and the treatment of any underlying diseases. The outcome of dogs with CVA depends on the size of the lesion, the location of the lesion and the severity of the clinical signs. Many cases of cerebral infarctions can improve dramatically over a few days to weeks; however, these cases are at risk of multiple events. Intraparenchymal hemorrhage may also cause reversible signs but the severity of both the clinical signs and the underlying diseases may often be more severe.
Classes & causes of cerebral infarction.
|
Class of Infarction |
Causes of Infarction |
|
(i) Arterial Obstruction |
|
|
--Thrombosis |
Atherosclerosis (Hypothyroidism / hyperlipoproteinemia / idiopathic)
Extension of CNS infection |
|
--Embolism |
Sepsis
Neoplasia
Dirofilaria immitis
Heart disease |
|
(ii) Arterial Occlusion |
Vasculitis
Arteriosclerosis |
|
(iii) Venous Thrombosis |
Inflammation
Neoplasia |
Classes & causes of cerebral hemorrhage.
|
Class of Hemorrhage |
Cause of Hemorrhage |
|
(i) Primary |
Hypertension |
|
(ii) Secondary |
Hemorrhagic infarction·
Cerebral amyloid angiopathy
Vascular malformation
Neoplasia
Vasculitis
Coagulopathy |
Time course of evolving infarction on computed tomography.
|
Time after Infarction |
CT Characteristics |
|
0-24 hours |
Normal or subtle hypodensity +/- sulca effacement |
|
1-7 days |
Mass effect (peaks at 3-4 days) |
|
Days to months / years |
Hypodensity |
|
1-8 weeks |
Contrast enhancement |
|
Weeks to years |
Atrophy |
Acute infarction: conventional magnetic resonance findings.
1. Lesion in arterial distribution
2. High intensity on proton density / fluid attenuated inversion
3. Gyri swollen, sulci effaced
4. Subcortical white matter hypointensity
5. Intravascular contrast enhancement
Physiologic factors influencing magnetic resonance appearance of hematomas.
 Age of hemorrhage
Age of hemorrhage
Site of hemorrhage
Size of hemorrhage
Local partial pressure of oxygen
Local pH
Hematocrit
Blood-brain barrier integrity
Presence of underlying lesion
Physiologic factors influencing magnetic resonance appearance of hematomas
Effect of age of hematoma on its magnetic resonance imaging characteristics.
|
Biochemical Form |
Clinical Stage |
Approximate Time of Appearance |
Intensity* on T1-weighted Image |
Intensity* on T2-weighted Image |
|
OxyHb in RBCs |
Peracute |
Immediate to first several hours |
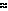
|
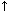
|
|
DeoxyHb in RBCs |
Acute |
Hours to days |
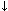 |
|
|
MetHb in RBCs |
Subacute |
First several days |
|
|
|
Extracellular MetHb |
Subacute to Chronic |
Days to months |
|
|
|
Ferritin & Hemosiderin |
Chronic |
Days to indefinite |
|
|
* Signal intensity is relative to normal brain parenchyma
References
1. Frankhauser R, Luginbuhl H, McGrath JT. Cerebrovascular disease in various animal species. Ann N Y Acad Sci 1965; 127: 817-859.
2. DeLahunta A, ed. Veterinary Neuroanatomy and Clinical Neurology. Philadelphia: W B Saunders Co. 1983: 130-155.
3. Garcia JH. The evolution of brain infarcts: a review. J Neuropathol Exp Neurol 1992; 51: 387-393.
4. Kalimo H, Kaste M, Haltia M. Vascular diseases. In: Graham DI, Lantos PL, eds. Greenfield's neuropathology. London: Arnold, 2002: 281-355.
5. Thomas WB. Cerebrovascular disease. Vet Clin N Am: Small Anim Pract 1996; 26: 925-943.
6. Summers BA, Cummings JF, de Lahunta A. eds. Veterinary Neuropathology. St. Louis: Mosby. 1995: 208-350.
7. Marks MP. Cerebral ischemia and infarction. In: Atlas SW, ed. Magnetic imaging of the brain and spine. Philadelphia: Lippincott Williams &Wilkins 2002: 919-979.
8. Toole JF, Burrow DD. Pathophysiology and clinical evaluation of ischemic vascular disease. In: Youmans JR, ed: Neurological surgery. Philadelphia: WB Saunders Co 1990: 1463-1515.
9. Wagner KR, Xi G, Hua Y, et al. Lobar intracerebral hemorrhage model in pigs: rapid edema development in perihematomal white matter. Stroke 1996; 27: 490-497.
10. Zazulia AR, Diringer MN, Derdeyn CP, Powers WJ. Progression of mass effect after intracerebral hemorrhage. Stroke 1999; 30: 1167-1173.
11. Wagner KR, Xi G, Hua Y, Kleinholz M, de Courten-Myers GM, Myers RE. Early metabolic alterations in edematous perihematomal brain regions following experimental intracerebral hemorrhage. J Neurosurg 1998; 88: 1058-1065.
12. Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. N Engl J Med 2001; 344: 1450-1460.
13. Castel JP, Kissel P. Spontaneous intracerebral and infratentorial hemorrhage. In: Youmans JR, ed: Neurological surgery. Philadelphia: WB Saunders Co 1990: 1890-1917.
14. Dukes J. Hypertension: A review of the mechanisms, manifestations, and management. J Small Anim Pract 1992; 33: 119-129.
15. Kazui S, Naritomi H, Yamamoto H, Sawada T, Yamaguchi T. Enlargement of spontaneous intracerebral hemorrhage. Incidence and time course. Stroke 1996; 27: 1783-1787.
16. Shores A, Cooper TG, Gartrell CL, et al. Clinical characteristics of cerebrovascular disease in small animals. In, Proceedings of the 9th American College of Veterinary Internal Medicine Forum 1991: 777-778.
17. Inoue Y, Takemoto K, Miyamoto T, et al. Sequential computed tomography scans in acute cerebral infarction. Radiology 1980; 135: 655-662.
18. Brant-Zawadzki M, Periera B, Weinstein P, et al. MR imaging of acute experimental ischemia in cats.Am J Neuroradiol 1986; 7: 7-11.
19. Grossman RI. Intracranial hemorrhage. In, Latchaw RE, ed. MR and CT imaging of the head, neck, and spine. St. Louis: Mosby Year-Book, 1991: 171-202.
20. Atlas SW, Thulborn KR. Intracranial hemorrhage. In: Atlas SW, ed. Magnetic imaging of the brain and spine. Philadelphia: Lippincott Williams &Wilkins, 2002: 773-832.